Research
Our research focuses on which genetic variations drive clonal hematopoiesis and how intrinsic and extrinsic factors regulate the evolution of hematopoietic clones.
We develop novel computational methods and apply existing methods to uncover biology underlying CH and leukemia development. Our research is facilitated by large-scale population and biobank cohorts with genotype and phenotype data (e.g., UK Biobank) and multi-omic datasets. Our projects are collaborative involving computational, experimental, and clinical scientists in various countries.
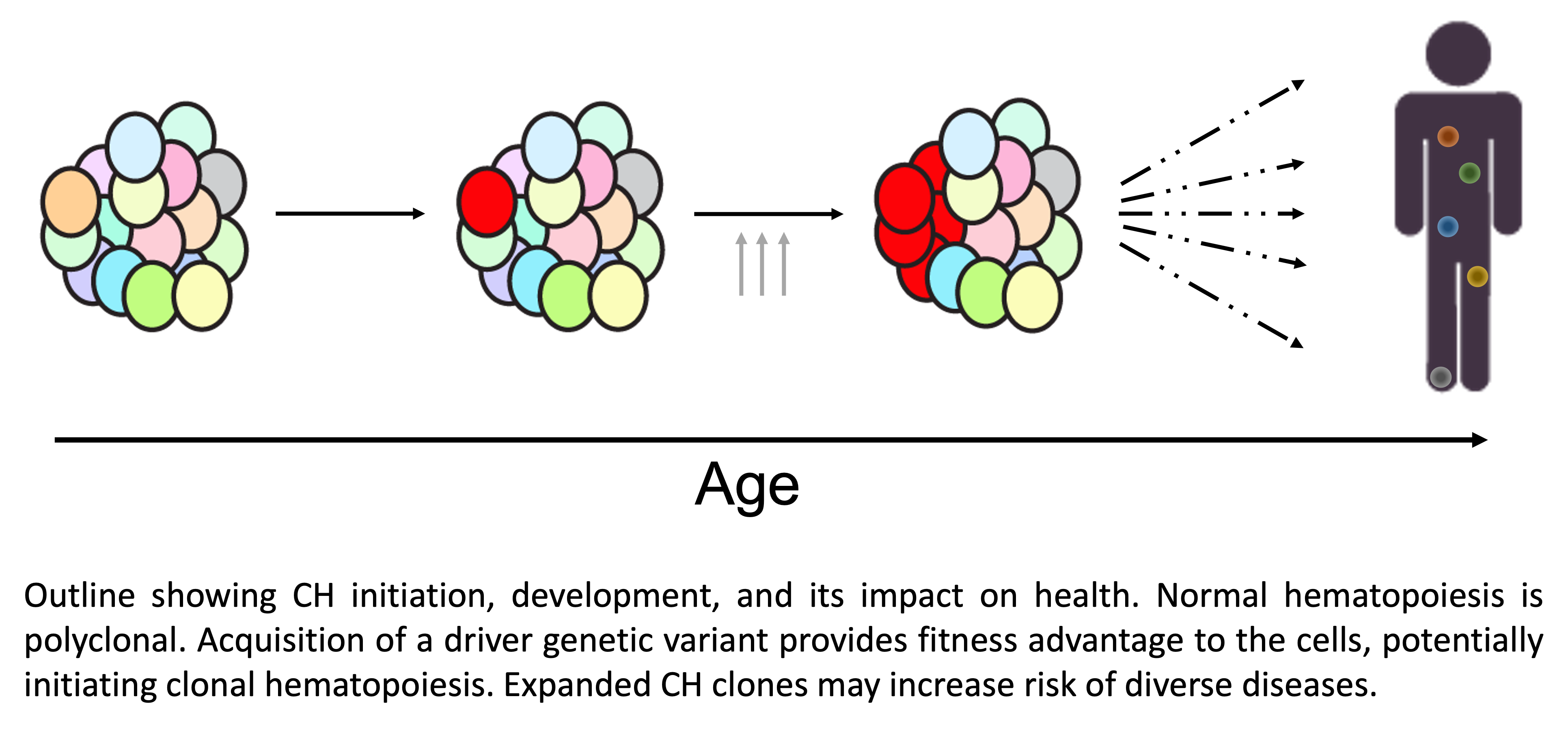
Origin and evolution of clonal hematopoiesis (CH)
In this project, we aim to map the landscape of driver genetic variations that give rise to CH. To date, genetic variations within protein coding genes and copy number alterations have been identified as drivers of CH. Recently available whole-genome sequencing datasets enable the analysis of genetic variations across the whole genome and detection of non-coding drivers of CH.
To understand the mechanisms of clonal evolution, we study how genetic and non-genetic factors influence CH development. Almost everyone acquires multiple driver genetic variations asssociated with CH. However, only a fraction of the mutated cells give rise to CH, which suggests strong selection pressure regulating the clonal evolution. The germline genetic background, combinations of somatic genetic variations as well as exposures to mutagenic factors may regulate clonal evolution.
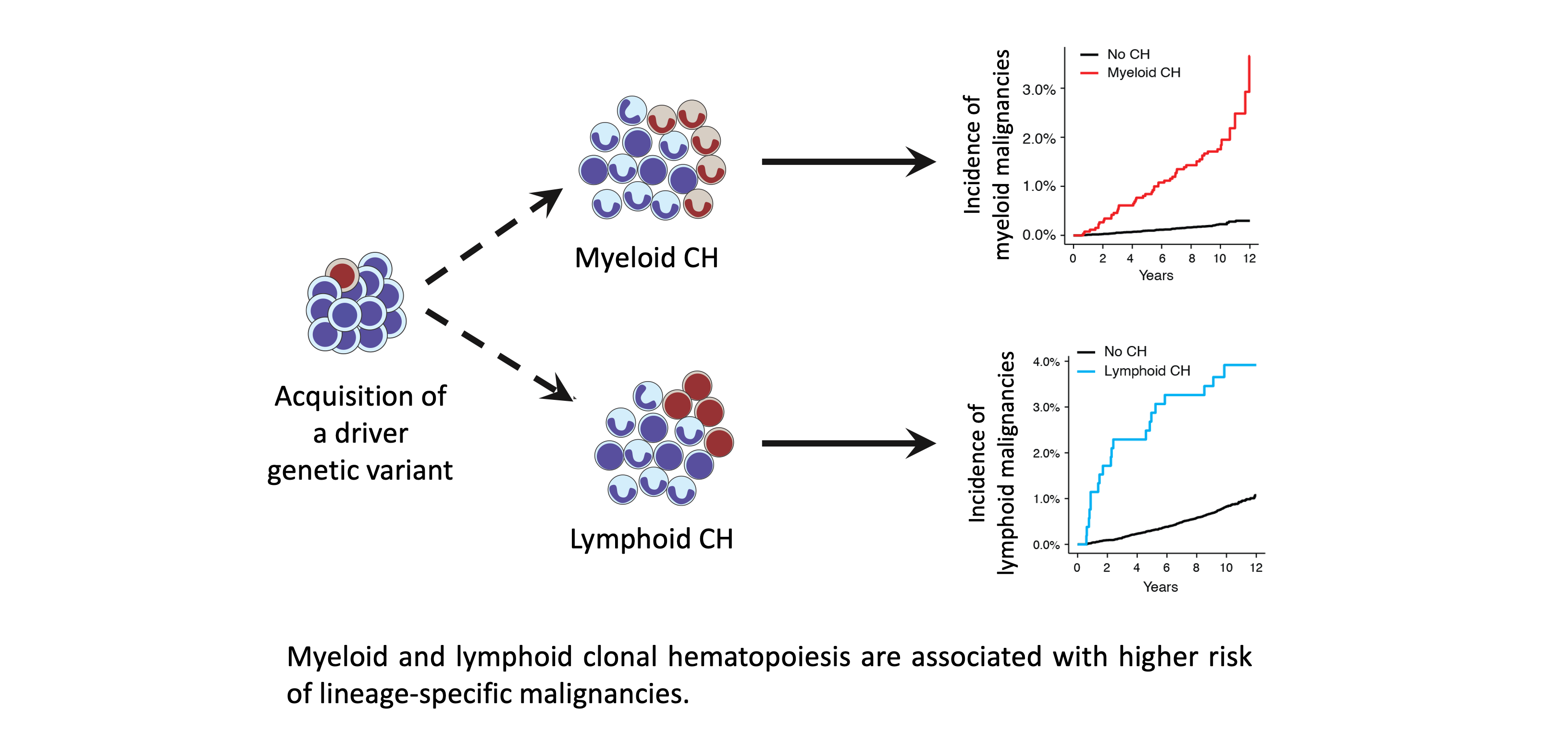
Impact of clonal hematopoiesis on health and disease
CH has been associated with increased risk of hematologic malignancies and a wide range of non-hematologic diseases. However, stratifying the risk associated with CH is challenging. Recently, we distinguished between myeloid and lymphoid which stratified the risk of developing myeloid vs lymphoid malignancies (Niroula et al., Nature Medicine 2021). We aim to identify markers that allow stratification of CH-mediated risk.
Selected publications
Distinction of lymphoid and myeloid clonal hematopoiesis. Niroula A, Sekar A, Murakami MA, et al. Nat Med (2021). doi: doi.org/10.1038/s41591-021-01521-4
Functional dissection of inherited non-coding variation influencing multiple myeloma risk. Ajore R, Niroula A, Pertesi M, et al. Nat Comms (2022). doi: doi.org/10.1038/s41467-021-27666-x.
MPRAscore: robust and non-parametric analysis of massively parallel reporter assays. Ajore R, Niroula A, Nilsson B. Bioinformatics (2019). doi: doi.org/10.1093/bioinformatics/btz591.